Machine learning is a hot topic, but like so many hot topics there is hype and confusion about what it is, and what it can (and can’t) do. Machine learning, which is a subset of artificial intelligence, can help experimental researchers achieve results they could not otherwise obtain quickly and easily. Machine learning models are trained on (ideally) thousands of data points to ‘learn’ the complex relationships between the data points and properties of interest, and are sometimes described as not being easily interpretable by humans (the so-called ‘black box’), although this is changing.
Deep learning is a subset of machine learning. Although this powerful technique is gaining popularity, deep learning is not always the best tool for the job. Like any research project in chemistry, the optimal machine learning technique depends on what you’re trying to achieve, and what information is available to you.
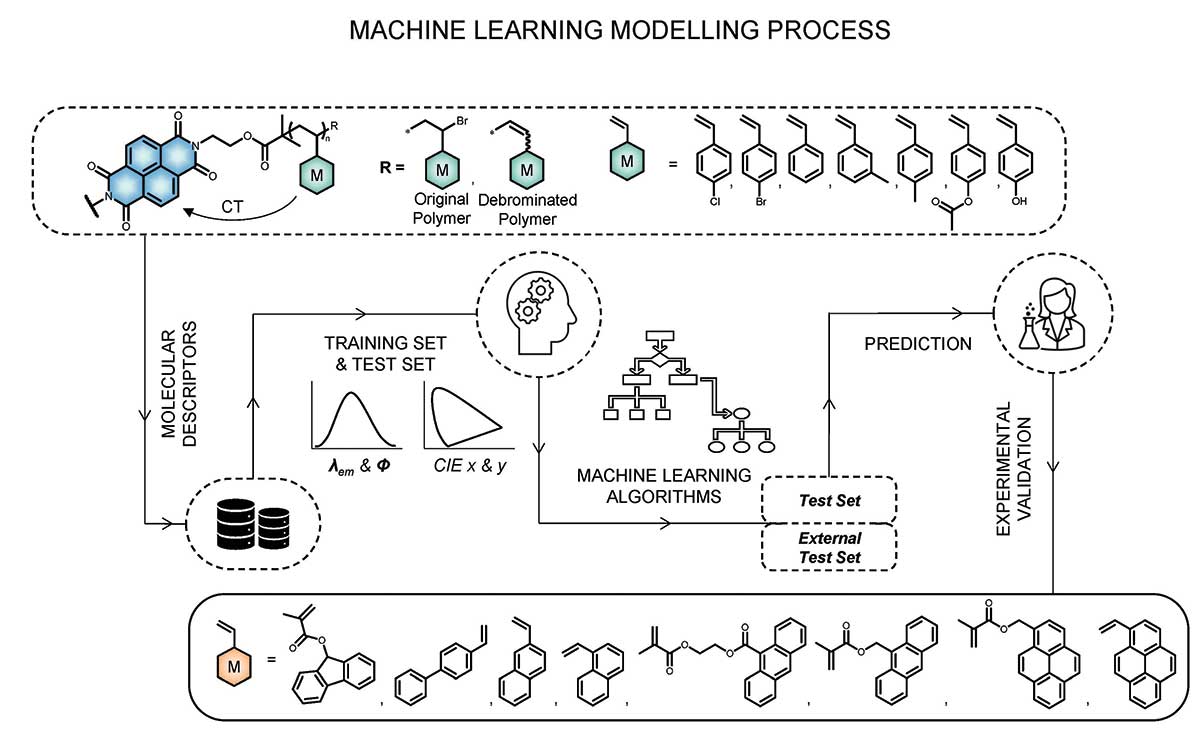
A library of polymer systems
In our efforts to achieve full-colour tuneability in fluorescent polymers, our experimental collaborators developed a library of 71 polymer systems where the number and chemical composition of the electron donor monomers were varied, but the electron-accepting core remained constant. For each system, properties such as colour and intensity were measured. Simply by trial and error, our colleagues were able to produce a variety of colours, but the goal was to be able to quickly produce polymers of any desired colour. This is where machine learning was invaluable. Machine learning models trained on the chemical composition and number of monomers in the polymer can predict the colour of the resultant polymer. Because machine learning can only make models out of numerical data, the colour of each system was described using CIE 1931 chromaticity coordinates, where the observed colour is translated into Cartesian x and y numerical values.
Choosing a machine learning model
The next step was to decide what type of machine learning model to use. Machine learning can be further broken down into linear and non-linear methods (although it’s a bit more nuanced than that). One of the most straightforward linear methods is multiple linear regression, where the equation y = mx + b is repeated for multiple x variables x1, x2, x3 and so on (also known as descriptors or features), and the algorithm solves for the constants b, m1, m2, m3 and so on. Once the model is optimised, the m values can tell you how strongly each descriptor contributes to the final property, and whether the effect of each descriptor is positive or negative. This ability to interpret the model and understand the contribution from each descriptor is one of the main advantages of multiple linear regression. One of the main disadvantages is that it only works when the relationship between the descriptors and the property of interest is linear.
Artificial neural networks are some of the most popular non-linear machine learning methods. They have been compared to the human brain in how they process and transfer information. In contrast to deep learning (multiple hidden layers containing large numbers of neurons that require very large training sets), shallow neural networks (a single hidden layer with few neurons) are able to generate robust models with a relatively small number of data points. The main limitation of these models is not so much their modelling accuracy but their small domains of applicability. In other words, they are not as good at predicting properties of things that are substantially different from the data set that was used to train the model. While artificial neural networks have the advantage of being able to determine non-linear relationships between descriptors and the property of interest, the contribution of each descriptor is a local rather than global property for non-linear models. This means that the magnitude and direction (positive or negative) of the contribution depends on where on the complex, multidimensional response surface the importance is assessed, and this is not easily obtained from the model. Simply put, artificial neural networks are good at predicting properties based on descriptors, but it is difficult to interpret how they achieved this prediction (the black box).
Both linear and non-linear machine learning methods require descriptors. Descriptors are mathematical entities that encode chemical and physical properties of materials. There are literally thousands of possible descriptors for each molecule or material, and most are not particularly informative. Unless the set of descriptors is reduced to those containing the most relevant information, models will be overfitted and degraded by the presence of uninformative descriptors (noise). A very efficient way to select the best subset of relevant descriptors that avoids the possibility of chance correlation between descriptors is the use of sparse feature selection methods that set less relevant descriptors identically to zero, improving model predictivity and interpretability.
Predicting promising polymers
In our models of photoluminescent properties of fluorescent polymers, our aim was to use molecular descriptors that are both efficient and chemically interpretable to describe the emission wavelength, quantum yield and CIE x y coordinates that define the colour, to provide guidance for the synthesis of new polymers of interest. We deliberately avoided using descriptors based on experimentally determined properties or derived from computationally expensive quantum chemical calculations, because they are unsuitable for predicting properties of materials not yet synthesised, or are very resource demanding. Instead, we chose the smallest number of simple descriptors that could produce a reliable model and were easily determined without synthesis or computationally expensive calculations, such as number of monomers in the polymer, type of monomer based on percentage of carbon or halogen atoms, and nature of the terminating group of the polymer.
We used both multiple linear regression and artificial neural networks to create our models and, in both cases, divided the experimental data set of 71 polymers into a training set of 57 and a test set of 14 polymers. It is standard practice in machine learning to divide the data set this way and use about 80% for the training set and 20% for the test set, in order to ensure that the model is not overfitted to the training set and can still predict the properties of materials it wasn’t trained on. While artificial neural networks produced slightly better models, multiple linear regression models allowed us to interpret how the characteristics of the polymers, as given by the descriptors, affected each property of interest.
Based on the outcomes from machine learning, our experimental collaborators synthesised and characterised new polymers from an additional eight monomers not previously used, and we used this data as an external test set to further validate our models. The properties of seven out of eight were predicted well. The eighth monomer had a significantly different structure from the monomers used in the model training sets, which emphasises that when the data set is small, machine learning often cannot be used to predict the properties of materials that are substantially different from those in the data set. The solution, of course, is that the new materials can then be added to the data set and used to develop new and better models (called active learning).
In general, to identify functional polymers with targeted performance, it is very time consuming and expensive to synthesise and characterise a large library of polymers one by one, considering all possible combinations of monomers, chain lengths, end group functionalisation, and so on. Using machine learning techniques, experimental scientists can greatly save time and resources by avoiding the experimental screening process and only synthesising the polymers predicted by machine learning to be promising. When there is a need to select polymer structures from a large pool of candidates, the machine learning models would play a pivotal role in reducing the working load for time-consuming and expensive synthesis.
Still, there are some considerations for experimental researchers to keep in mind if they are interested in using machine learning to optimise their research. For those not familiar with machine learning, it is important to emphasise that the property of interest is the output, not the input, of the model. If, for example, we wanted to produce a polymer that is green, the machine learning model can’t necessarily tell us what to synthesise. Instead, we can input thousands of possible combinations of monomers, chain lengths, and end group functionalisations as descriptors, and the models will predict in a matter of seconds which of those are most likely to be green. In the case of multiple linear regression, the interpretability of the model allows us to narrow down the choice of possible combinations most likely to be green.
Another important consideration is that machine learning doesn’t always work. This could be because of the data set, particularly if the data set is compiled from the work of different experimental groups, or simply because there is no clear relationship between the descriptors chosen and the property of interest. In both cases, the solution can be the same: use a larger data set produced by a single group and, if possible, try different descriptors.
Machine learning and the Sustainable Development Goals
Our world is rapidly changing, and in materials discovery and development we can’t afford to wait 40 years between the initial innovation and full commercial utilisation. In the future, machine learning will be essential to achieve the Sustainable Development Goals outlined by the United Nations, particularly affordable and clean energy, industry, innovation and infrastructure, and climate action.
In collaboration with experimental colleagues across Australia and beyond, we have already developed machine learning models for organic and inorganic solar cells that have improved their efficiency and provided non-intuitive guidelines for further optimisation, and we are currently working on developing and optimising carbon capture technologies for commercial use. In addition to materials composition, machine learning can provide models and predict other variables such as temperature, duration and cost/efficiency ratios that will factor into the commercial implementation of the materials and systems. Machine learning is undoubtedly becoming an essential component of materials discovery, design and implementation, and has enabled us to achieve outcomes more quickly and efficiently than ever possible previously.