Fundamental understanding of specific disease biology can pinpoint proteins that may be involved in a disease of interest. For example, elevated levels of protein X are associated with an increased risk of cancer. The next step in the target validation process would traditionally involve the generation of genetically modified animals in which the gene coding for protein X has been removed in utero, thus generating a ‘knock-out’ animal model. The knock-out animal no longer produces protein X and subsequently lacks that protein’s biological processes. We then compare the knock-out animal with its wild-type cousin in models of our disease of interest to assess the impact of removing protein X.
If the knock-out animal shows an improvement in the progression, severity or in some cases immunity of disease, then X becomes a protein of interest (POI). We also class the response of this animal in the disease models as the knock-out phenotype.
The use of RNAi and CRISPR techniques has expanded our ability to generate these knock-out phenotypes both in mature animals and in individual cells. The advantage of these techniques is that removal of our POI occurs in a mature animal, thus removing any adaptions that may occur in the maturation of a traditional knock-out animal.
As a medicinal chemist, I have spent over half my professional life trying to recapitulate the phenotype observed in these genetically modified knock-out animals by using small molecules that block the function of the POI in the wild type system. Using well-established medicinal chemistry techniques, I can design a small molecule that will be able to interact with our POI with sufficient potency and efficacy, and have the appropriate properties to enable inhibition of the function of the protein in vivo.
Unfortunately, this small molecule approach does not always fully imitate what happens in the POI knock-out animal. One reason for this difference is that while the RNAi and CRISPR gene editing techniques remove the POI in its entirety, my small molecule can only block its function, which could be both partial and transient in nature.
To borrow from Donald Rumsfeld, there are ‘the unknown unknowns – the ones we don’t know we don’t know’. In our case, there may be unexpected impacts of removing the POI, which may not be related to its function. For example, is our POI involved with other processes unrelated to its perceived function and is it through this route that the required phenotype is generated? This is especially true of protein kinases for which there is an increasing body of literature describing a variety of their non-catalytic roles, such as scaffolding, allosteric regulation of other proteins and even protein–DNA interactions.
So, to come back to my first point – in some cases will the functional inhibition of the targeted POI ever replicate the phenotype observed in the traditional knock-out animal? We have already discussed the advantages of the CRISPR and RNAi biological techniques, so is it therefore possible to have a chemical equivalent to CRISPR? Can we use small molecule medicinal chemistry to remove the POI from a mature test animal and ultimately in the clinic?
Pioneering work from the laboratory of Craig Crews, Yale University, US, may now provide a means of removing the POI by small molecule techniques by hijacking the cell’s own garbage disposal system, the ubiquitin proteasome pathway. The 2004 Chemistry Nobel Prize-winning work of Ciechanover, Hershko, Rose and others has delineated the molecular mechanisms involved in the ubiquitin pathway of protein degradation and it is upon this work the Crews group has developed the concept of PROTACs.
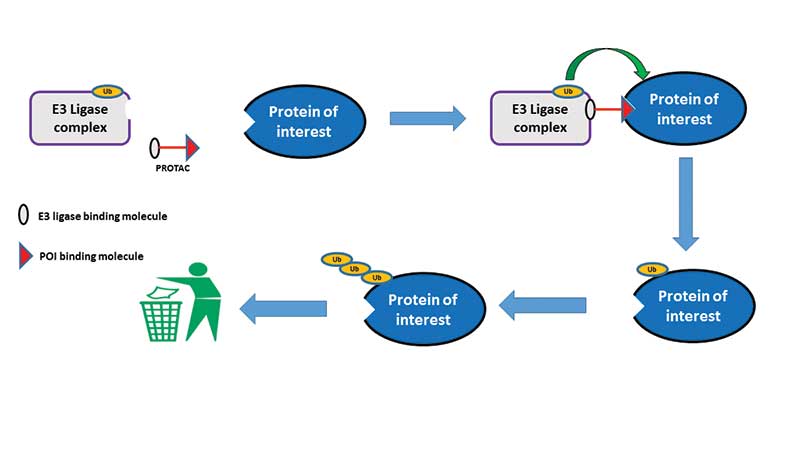
The degradation of a protein via the ubiquitin proteasome pathway involves a discrete two-step process – the first step requires the tagging of the protein with ubiquitin molecules and the second step recognises the specifically tagged protein and subsequently destroys the protein in the proteasome. The key step in the ubiquitination of the protein involves an enzyme called an E3 ligase. It is this E3 ligase that catalyses the transfer of the ubiquitin molecule from the ubiquitin-containing enzyme to an available lysine on the protein that is to be degraded.
The revolutionary approach from Crews and others was to design heterobifunctional molecules (molecules that are capable of binding contemporaneously to two different targets) that could bind both to the E3 ligase and to our POI. The heterobifunctional molecules are termed PROTACS (PROteolysis TArgeting Chimera).
It is the formation of this ternary complex, PROTAC/E3 ligase/POI, that is key for the degradation of our POI. The E3 ligase component of this tertiary complex then transfers the ubiquitin to our POI, which thus starts the process that eventually ends with the destruction of our POI in the proteasome (see scheme below). After the tagging of the POI with ubiquitin, the PROTAC is free to dissociate from the tertiary complex and is then able to interact with further molecules of our POI and E3 ligase. In this setting, the PROTAC is acting in catalytic mode since one molecule of PROTAC has the potential to degrade multiple copies of the protein.
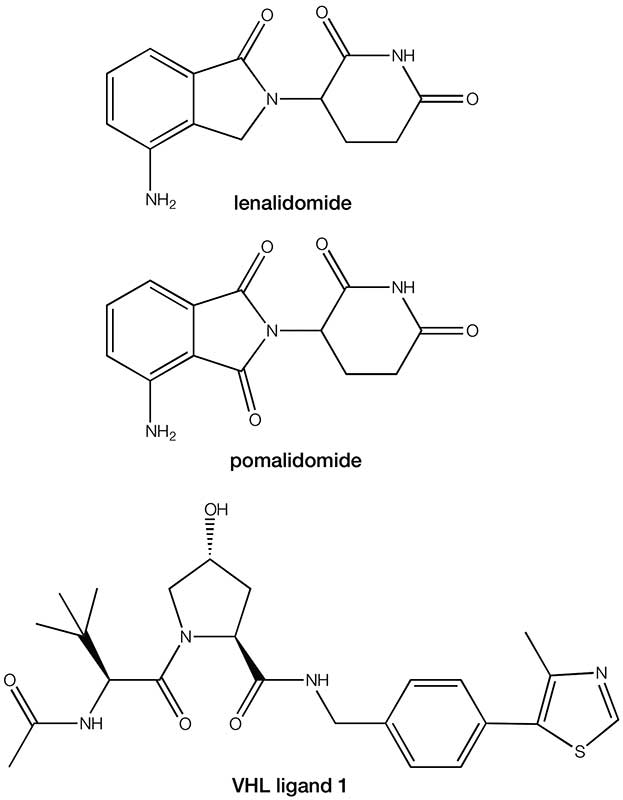
From traditional medicinal chemistry work, we already have discovered a plethora of small molecules that inhibit multiple POIs, so the key requirement in the development of this approach was to now identify suitable small molecules that could bind to the E3 ligase. The discovery that the immunomodulatory drugs lenalidomide and pomalodomide exert their therapeutic effect through binding to the E3 ligase cereblon provided the second part of the jigsaw. An alternative area of E3 ligase exploration has focused on the von Hippel-Lindau complex and its primary ligand hypoxia-inducible factor 1a (HIF-1a). Elegant structure activity work with HIF-1a has provided compound VHL ligand 1 as an additional ligand for the E3 ligase to lenalidomide and pomalodomide.
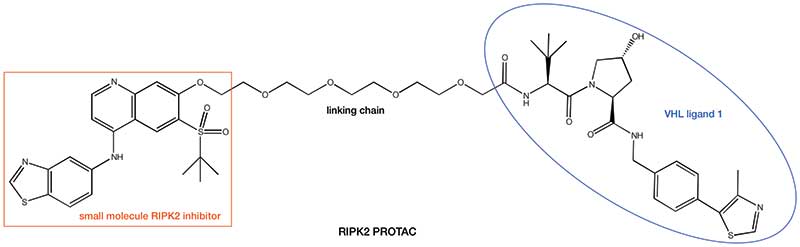
With two pieces of the PROTAC jigsaw now available, it just remains to find suitable methodology to connect them to each other. A significant number of small molecule drug discovery programs are driven by the use of X-ray crystallography studies to rationalise how small molecule ligands bind to their POIs. Analysis of this structural information helps to direct where to append a linking chain to our small molecule to avoid disruption of binding to the POI. The presence of the free nitrogen atom in lenalidomide and pomalodomide provides a suitable anchor point for the other end of the linking chain. In the case of the VHL ligand 1, unmasking the terminal amide provides another amine nitrogen atom suitable for using as a linkage point. Utilisation of this strategy arrives at a compound such as the RIPK2 PROTAC shown below. Work from the the laboratory of Alessio Ciulli, University of Dundee, UK, another of the other pioneers of the PROTAC approach, has recently solved the first ternary complex crystal structures, which aid in the design of the linking chain.
A recent edition of the Journal of Medicinal Chemistry highlights the number of protein targets that are amenable to this approach, including kinases (such as the RIPK2 example), epigenetic modifiers (bromodomains and sirtuins) and nuclear hormone receptors (oestrogen and androgen receptors).
Two further features of the PROTAC that offer exciting possibilities in the drug discovery world are the catalytic nature of the PROTAC process and the fact that the small molecule that I design now only needs to bind with our POI; no longer would my small molecule need to inhibit the POI.
The catalytic nature of the PROTAC has led to what Crews has termed ‘event-driven pharmacology’ rather than the classical exposure-driven pharmacology. In the traditional drug design, the efficacy of the drug is determined by a combination of pharmacokinetics (exposure), pharmacodynamics and target engagement. With the PROTAC’s ability to eliminate the POI, the desired efficacy need not match the exposure but is actually driven by the re-synthesis rate of our POI.
With the PROTAC approach, we only have to design molecules that bind to our POI because the pharmacology is now driven by removal of the POI. This will increase the number of protein targets we as medicinal chemists can target since it is suggested that current therapies only target 13% (400 out of 3000 genes) of the therapeutic proteome (the proteome being the entire set of proteins expressed by an organism at a certain time).
On the basis of our current drug discovery paradigms, a molecule with the physical characteristics of a PROTAC owing to the conjugation of two effective protein binders (high molecular weight, high polar surface area and high logD) would be unlikely to demonstrate activity after oral dosing. However, studies from the Crews group and others have shown in vivo efficacy in animal models after their PROTACs were dosed orally.
These findings have been the basis of two fledgling biotech companies, Arvinas (a 2015 Fierce 15 company) and C4 Therapeutics (a 2016 Fierce 15 company), forming to take this technology into the clinic. In late 2017, Arvinas announced the progression of the first PROTACs into clinical trials – ARV-110 as an orally bioavailable degrader for the androgen receptor for the treatment of metastatic castration-resistant prostate cancer and ARV-378 as a PROTAC targeted against the oestrogen receptor for the treatment of metastatic breast cancer.
We are currently awaiting the read-outs from the current clinical trial using the CRISPR technology and from Arvinas to see if this chemistry-based technology will complement CRISPR.
PROTACs may also find a home in phenotypic screening. Exploiting cell-based phenotypic screening is an increasingly frequent strategy deployed in drug discovery. This approach can identify new protein targets with novel modes of action; however, these proteins are often poorly characterised, and lack readily identifiable enzymatic activity, tool ligands and biomarkers of target engagement. A recent report has used a PROTAC to probe against the poorly understood and non-catalytic target pirin. Pirin has no known enzymatic function in mammalian cells, no reported endogenous ligands and no described validated proximal biomarkers. Previously the team at the Institute of Cancer Research had used classical pull-down techniques to identify that the target of their small molecule was pirin. Switching to the PROTAC containing the small molecule, they were able to show degradation of their POI, pirin, and begin to understand the pharmacology associated with this target. Again, PROTACs are acting as the chemical equivalent of classical knock-out/down techniques in the understanding of the role of specific proteins in the cell.
The pioneering work from Crews and Ciulli can now provide PROTACs as an alternative to the biology-based CRISPR and RNAi techniques. It will be interesting to see how the PROTACs will stack-up against the CRISPR techniques. In the near future, the first read-outs will be available from the relative clinical trials. If the PROTACs are successful in the clinic, then the drug development route would follow the standard small molecule CMC pathway. I look forward to seeing if the PROTAC approach will complement or surpass the potential of CRISPR.