The 2017 Nobel Prize in Chemistry was jointly awarded to three researchers for their convergent work in cryo-electron microscopy. With breakthroughs as recently as 2013, this technology is already revolutionising the high-resolution structure determination of biomolecules in solution.Working separately over decades, Jacques Dubochet (University of Lausanne, Switzerland), Joachim Frank (Columbia University, New York) and Richard Henderson (MRC Laboratory of Molecular Biology, Cambridge, UK) developed critical aspects of this technology.
The first article in this series (February issue, p. 20) examined the basic technology and the fundamental problems that needed to be solved to make this analytical technique work. In this article, we will examine the individual approaches and contributions made by each of the researchers.
Jacques Dubochet
Swiss biophysicist Dr Jacques Dubochet commenced his physics studies in 1962 at what is now called the École Polytechnique Fédérale de Lausanne. Graduating in 1967, he went on to obtain a Certificate of Molecular Biology at the University of Geneva, then began his study in the electron microscopy (EM) of DNA. He completed his thesis in biophysics in 1973, through the University of Geneva and University of Basel.
The physical conditions of EM are punishing for biological samples. For a long time, it was widely considered that the dehydrating effects of the vacuum and the radiation-induced damage to the materials from the intensive, high-energy electron beams required to achieve contrast against the background made EM impractical for any but ‘dead’ samples.
Staining was extensively used from the 1940s to increase the differentiation against background. Biological samples were embedded in a thin amorphous film of a heavy metal salt, generating a ‘cast’ around the object. The cast would scatter electrons more strongly than the encapsulated material (increasing contrast, and creating a negative image), was more resistant to electron damage, and would retard the vaporisation of water from the sample. This was sufficient to provide information about the morphology of bacteria and viruses, but for single molecules or molecular complexes it only gave information about the envelope of the covered particles, and the resolution was limited to the granularity of the stain.
From the mid-1970s, Richard Henderson (discussed below) and Nigel Unwin developed a new preparation method to enable EM studies of unstained protein crystals at room temperature. They replaced water with a glucose solution to preserve the samples under vacuum, and used this method to study intact protein crystals.
However, it was Dubochet who generated the critical breakthrough in enabling analysis of liquid samples. Back in the 1950s, Humberto Fernández-Morán had already begun exploring the possibility of freezing samples and preparing thin sections for cryo-EM. It was expected that extreme cooling would both reduce water evaporation and protect biological samples from damage. But crystalline ice strongly diffracts electrons, and its formation may change the specimen structure.
Dubochet’s technique involved the rapid freezing of samples using ethane (which was in turn cooled by liquid nitrogen). When cooled in this way, water solidifies in its liquid form as a glassy material called ‘vitrified water’ – which is now believed to be the dominant form of water throughout the universe. In this state, the supercooled liquid water preserves the biomolecules in their natural shape, even under vacuum.
It is notable that this work built on earlier studies by Kenneth Taylor and Robert Glaeser, but the technology at the time did not allow studies below –120°C, which is above the transition temperature from vitrified water to crystalline ice. Nonetheless, Taylor and Glaeser offered important contributions for specimen handling at cryogenic temperatures.
So in 1981, Dubochet and Alasdair McDowall presented a method for the formation of thin-film non-crystalline samples on a specimen grid, ready for EM analysis. The sample was sprayed on a carbon film mounted on a grid, then rapidly immersed in liquid ethane or propane at about –190°C. At cryogenic temperatures, the vitrified water allowed nearly uniform absorption of electrons, yielding impressive contrast. These samples could be maintained for long periods provided temperatures were maintained below –160°C.
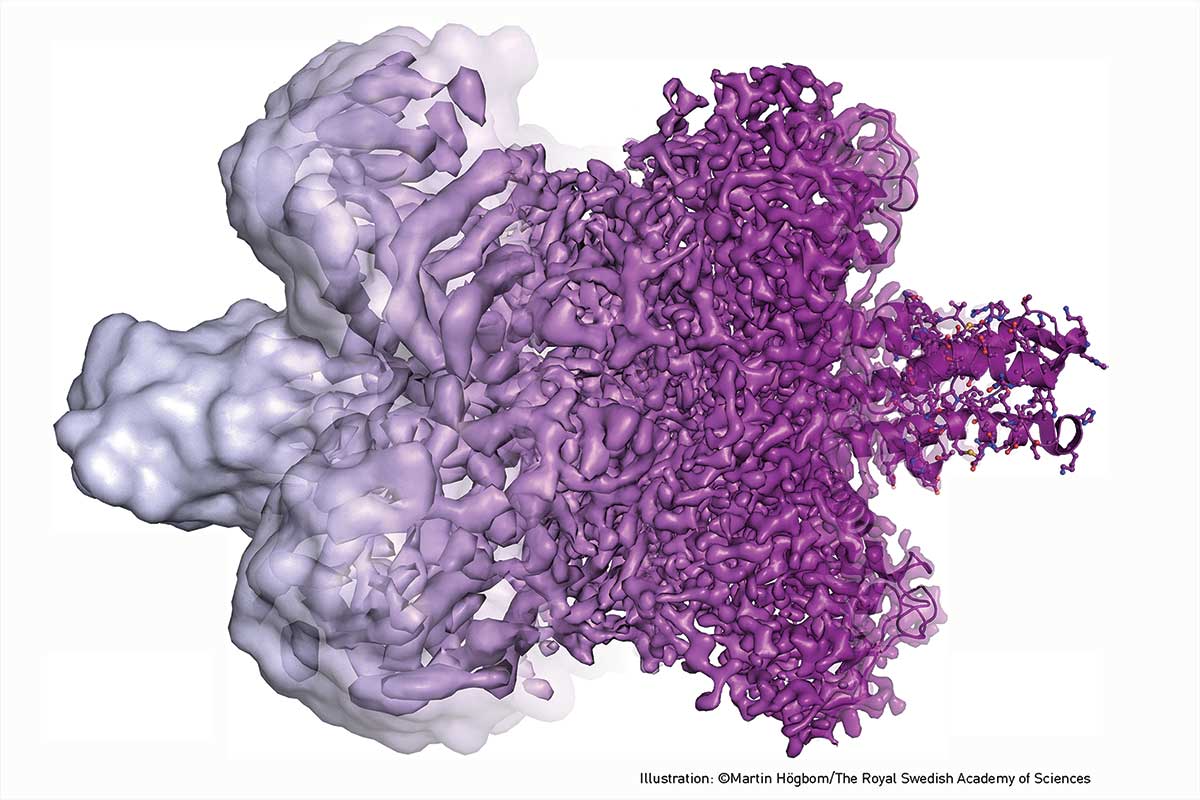
With this technique now widely adopted, samples can be made sufficiently thin to allow rapid vitrification, freezing movement for ‘snapshot’ of activity at a given moment in time. By careful collection of multiple samples, ‘movies’ of action can be constructed to develop detailed understanding of the activity in biological processes.
But cryo-EM is not only about static structures. Because sample preparation for cryo-EM involves instant cooling of a solution, the contents of the solution can be systematically varied; integral membrane proteins may be studied in a near-native environment; and the particles may be trapped in structural sub-states or even in action, for example, while an enzyme catalyses a chemical reaction. The data may offer functional information: structural changes may be monitored and free-energy landscapes determined. (bit.ly/2y8zInm)
Aged 65, Dubochet is now retired, and is an honorary professor at the University of Lausanne.
Joachim Frank
Born in Germany, Dr Joachim Frank is a Professor of Biochemistry and Molecular Biophysics and of Biological Sciences at Columbia University, and Distinguished Professor of the State University of New York at Albany. He received his diploma in physics at the University of Munich, and conducted his doctoral research at the Max Planck Institute for Biochemistry, Martinsried and at the Technical University of Munich.
Frank’s postdoctoral research at the Cavendish Laboratory in Cambridge, UK, and in the USA, addressed problems with electron optics and image processing. He joined the Wadsworth Center in Albany, New York, in 1975, and developed the single-particle reconstruction approach.
Biological samples are rarely structurally uniform and may contain impurities. So a major problem with studies of unstained, non-crystalline, asymmetrical and randomly oriented particles in solution is, according to a 1975 paper by Frank, ‘the alignment of features that are only faintly visible on a noisy background’. (bit.ly/2AAr1VK)
Frank and colleagues began studies of this issue in the 1970s and, by using studies of negatively stained glutamine synthetase, determined that it would be possible to average images of many sensitive particles to eventually construct high-resolution data, provided that the particles were uniform.
In 1981, Frank and Marin van Heel then extended this to present a method for non-uniform particles, which sorted the particle images into classes based on their orientation and structural features. By applying multivariate statistical analysis to sort the vector clusters, these clusters were then each assumed to represent a 2D projection of a particle with specific orientation and structural properties. It was then assumed that the particles within each cluster were sufficiently similar to allow averaging, increasing the signal to noise ratio.
In translating these 2D structures into 3D visualisations, Frank developed critical mathematical tools. Most particularly, Frank and Michael Radermacher’s Random Conical Tilt method combined the earlier general ideas with the application of a tomographic conical tilt series. The tools have been gathered into a suite of computer programs called SPIDER and made available for ready use by the scientific community.
In a 1976 issue of Research News (bit.ly/2BgYIeU), US biochemist Arthur Robinson commented on Frank’s work ‘exploring theoretical methods for averaging data from arrays of identical objects that are not periodic’: ‘If such methods were to be perfected, then, in the words of one scientist, the sky would be the limit’. A 2015 post by Frank concludes: ‘Well, we have reached the sky now, but the history of the single-particle techniques that brought us here goes back quite a few years’. (bit.ly/2BXVauL)
Frank is a Fellow of the American Association for the Advancement of Science and the Biophysical Society. He was elected to the National Academy of Sciences in 2006, as well as the American Academy of Arts and Sciences, and the American Academy for Microbiology. In 2014, he was awarded the Franklin Medal in Life Science.
Richard Henderson
Dr Richard Henderson initially studied physics at Edinburgh University, but switched to molecular biology at age 21. As a research student, he worked for the MRC Laboratory of Molecular Biology (LMB) at Cambridge, UK, where David Blow’s team was using
X-ray crystallography to prepare one of the first protein structure characterisations (for the enzyme chymotrypsin).
As a Helen Hay Whitney postdoctoral fellow at Yale, he developed an interest in the structure of membrane proteins. Then after returning to LMB in 1973, he commenced important work in collaboration with Nigel Unwin, developing new sample preparation methods for electron microscopy.
Henderson initially turned to EM after X-ray crystallography failed in the imaging of proteins embedded in the membrane surrounding the cell. Such proteins are often difficult to work with, because when removed from the membrane they clump up into a useless mass. In Henderson’s case, he simply couldn’t crystallise the selected protein for X-ray studies.
In an initial study (published in 1975), Henderson and colleagues simply worked with the enzyme while still embedded in its membrane. They were fortunate that the selected enzyme molecules happened to be regularly packed and oriented in the same direction on the membrane. The researchers prepared 2D projections of bateriorhodopsin crystals on the purple membrane, with images of the structures and diffraction patterns. They then used Fourier transformation of the structure images to calculate phases. In a subsequent study, they tilted the specimen to collect 2D projection images from multiple directions, and from these calculated low-resolution visualisations of the general structure of the protein by using methods developed by Aaron Klug (Nobel Prize for Chemistry, 1982) and colleagues. It turned out to be the best picture of a protein ever generated by an electron microscope, with a resolution of an impressive 0.7 nanometres.
Over subsequent years, multiple aspects of the technique improved, critically including both the lenses and the cryotechnology. As noted by Professor Peter Brzezinski of Stockholm University (bit.ly/2y8zInm): ‘The year 1990 marked a critical milestone when Henderson and colleagues showed for the first time that it is possible to obtain high-resolution structures of biomolecules using cryo-EM through averaging over many copies of the same object’.
In an interview following the prize announcement, Henderson called attention to the fact that the Nobel Prize can be awarded to at most three recipients, and graciously acknowledged that there were ‘others who’ve made strong contributions who didn’t quite cross the threshold’.
From dream to reality
The Swedish Academy of Sciences puts it succinctly (bit.ly/2AfEwpE):
Now the dream is reality, and we are facing an explosive development within biochemistry. There are a number of benefits that make cryo-EM so revolutionary: Dubochet’s vitrification method is relatively easy to use and requires a minimal sample size. Due to the rapid cooling process, biomolecules can be frozen mid-action and researchers can take image series that capture different parts of a process. This way, they produce ‘films’ that reveal how proteins move and interact with other molecules.
Such is the benefit of this new technology, that its range of applications is diverse. But cryo-EM is demonstrating its power, with significant practical outcomes already achieved and more still being conceived.