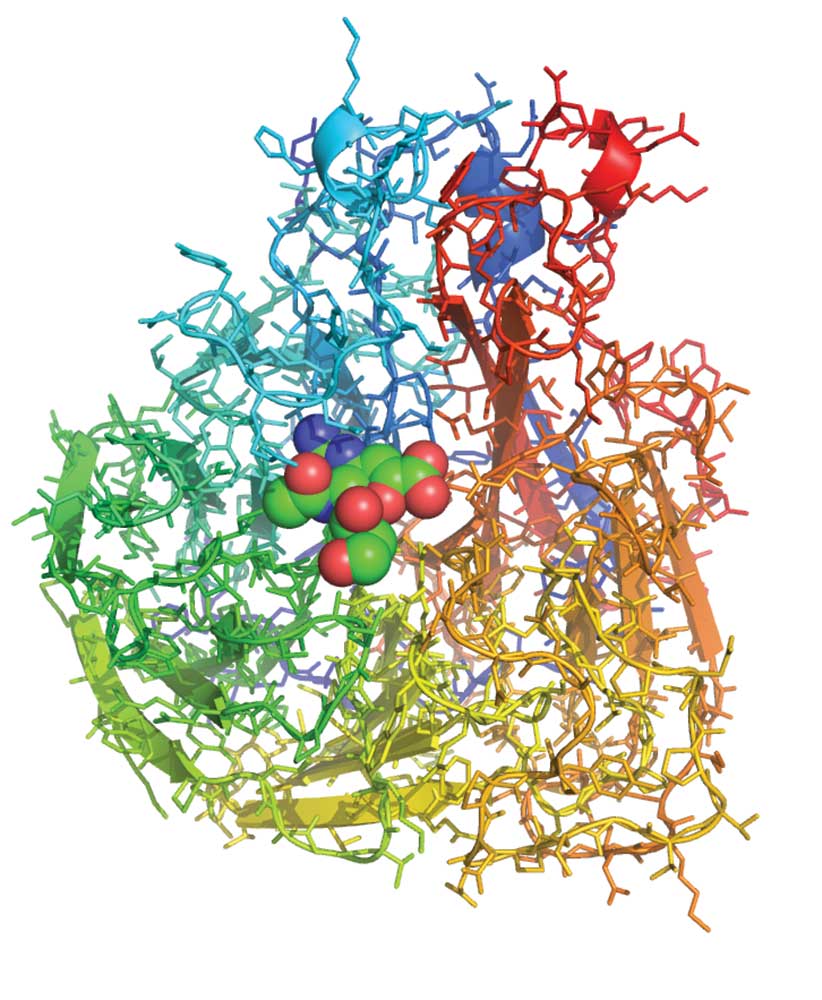
The cover of Nature on 5 May 1983 announced the description of the three-dimensional structure of the influenza virus protein neuraminidase. Jose Varghese, Graeme Laver and I used X-ray crystallography to pinpoint the position of more than 3000 non-hydrogen atoms in the structure, showing that the protein chain was folded up in a novel way, resembling a propeller. In the preceding months, others, including our CSIRO colleague Colin Ward, had worked out the sequence of the 400 or so amino acids within the protein, building up a linear picture of how one strain of the virus varied from another. These results, when laid out in three dimensions on the crystal structure, showed that amino acids subject to strain variation were distributed around the surface of the protein but those that were conserved between strains were concentrated in a pocket-shaped feature that we identified as the ‘active site’ of neuraminidase.
The role of the neuraminidase protein in the viral life cycle is to allow the release of progeny virions from an infected cell. Without a neuraminidase function, the newly made viruses remain tethered to the infected cell by an interaction between another viral protein – the haemagglutinin – and a receptor molecule on the infected cell surface. By clipping off this receptor, the neuraminidase liberates the viral progeny to go and infect other cells. The particular 3D arrangement of amino acids within the active site, recruited there from linearly distant positions in the sequence by the folding of the protein chain, provides the energy required to cut the chemical bond between the receptor molecule and the infected cell. Thus, it is unsurprising that those amino acids in the active site are preserved in all strains of virus; changes to them destroy the neuraminidase’s cutting action, without which the virus is not viable.
Between 1985 and 1990, supported by a start-up company (Biota Holdings Ltd), Varghese and I worked closely with a team of chemists, including Wen-Yang Wu and Mark von Itzstein, recruited for the purpose of designing and synthesising molecules that might disable the neuraminidase. The idea was that a compound that snugly fitted the neuraminidase active site and inhibited the protein’s activity would interrupt the viral life cycle and be effective against all strains of virus. In 1990, Biota licensed our discovery, zanamivir, to Glaxo (now GSK) to see if it could be developed into a drug. In 1993, Nature reported the results of our work, including a demonstration by Glaxo that the compound was effective in a ferret model of influenza if delivered intranasally. Three years later, competitors emerged with a close analogue of zanamivir, oseltamivir, that (unlike zanamivir) is membrane-permeable and enters the bloodstream if administered orally. This discovery was driven by the expectation that patients (and physicians) would prefer a drug in tablet form. (That has proven to be the case, even though a topically delivered drug that is unable to cross a membrane is effective because the neuraminidase is located on the outside of the virus and exerts its function when the virus is outside host cells.)
The first study of the efficacy of zanamivir in humans was performed by Fred Hayden at the University of Virginia, USA, and reported in 1996. Hayden’s result clearly showed that zanamivir was effective in reducing the viral burden in infected volunteers, but the effect would be marginal if the delay between infection and drug intervention exceeded two days. This result has much to do with the normal cycle of influenza infection, which typically resolves naturally after a week or so, having reached its peak 2–3 days after infection.
Clinical trials over the following years led to registration of both compounds in 1999, zanamivir formulated as Relenza and taken by oral inhalation and oseltamivir formulated as Tamiflu and taken orally. Recently (2014), both of the companies involved, GSK and Roche, have come under fire from the Cochrane Collaboration for selectively publishing results of these trials, some of which showed only marginal benefit in the median time to recovery for trial subjects. (The so-called ‘end-point’ of these trials is reached when a trial subject declares themselves well again.) It is arguable whether median time to recovery is the best end-point for these difficult clinical trials, which can only be performed when influenza is circulating in the community. Nevertheless, nothing less than full disclosure of clinical trial results should be required for drug registration. Cochrane, a group that monitors and reviews healthcare measures, argued that the benefits of the drugs did not justify expenditure by governments in stockpiling them for use in the event of an influenza pandemic.
Immediately following the Cochrane report came the PRIDE Study (University of Nottingham, UK) on the use of the neuraminidase inhibitors on some 30 000 patients hospitalised during the 2009 influenza pandemic. This study did not measure time to recovery but measured recovery itself. For this patient cohort, the odds of dying were halved if they received the drug within two days of the onset of symptoms compared to those whose treatment was delayed to five or more days. The estimated death toll from the 2009 pandemic is in excess of 200 000 and the study suggests that halving that number would have been achievable. Governments continue stockpiling neuraminidase inhibitors, though a debate with Cochrane continues over the PPRIDE study, now centred on methods of statistical analysis of data.
But, how has the claim of drug efficacy against all strains of virus played out? The devil really is in the detail.
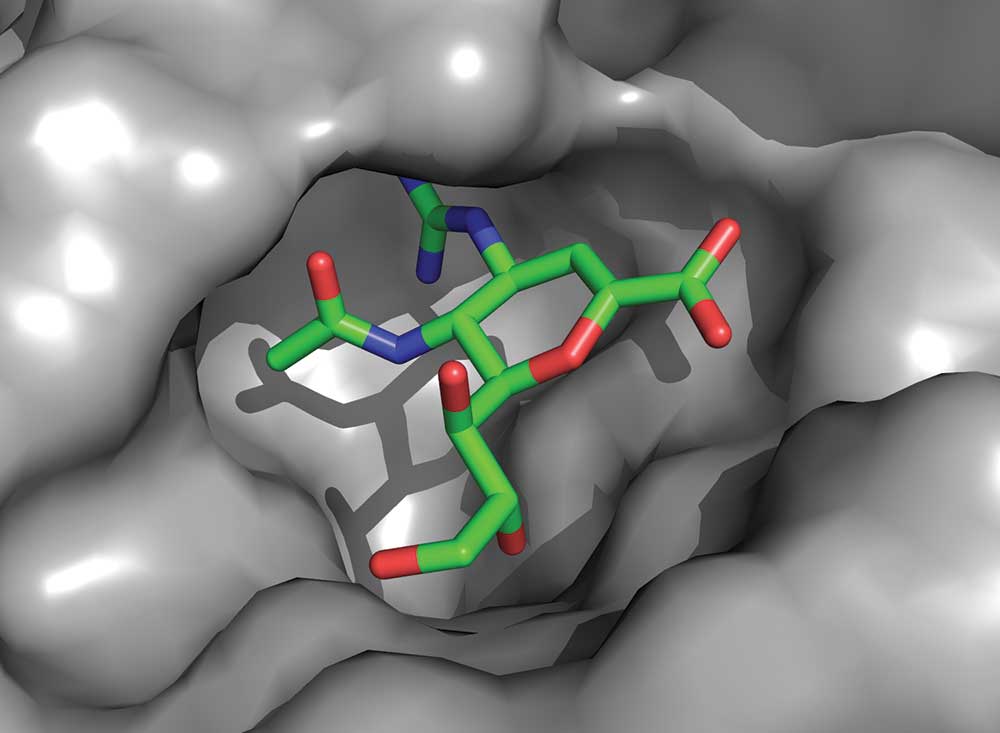
Our crystallographic experiments on neuraminidase were performed both with the protein alone and with the protein bound to the influenza receptor sialic acid. This showed that no changes occurred to the neuraminidase structure when it engaged the receptor for its cleavage. Zanamivir is very similar in structure to sialic acid. Only minor, though critical, changes were made to sialic acid to enhance the affinity for the protein and to generate the drug. The crystallography of zanamivir bound to the protein showed that it engaged only conserved amino acids in the active site and, like sialic acid, required no change to the protein structure for its binding.
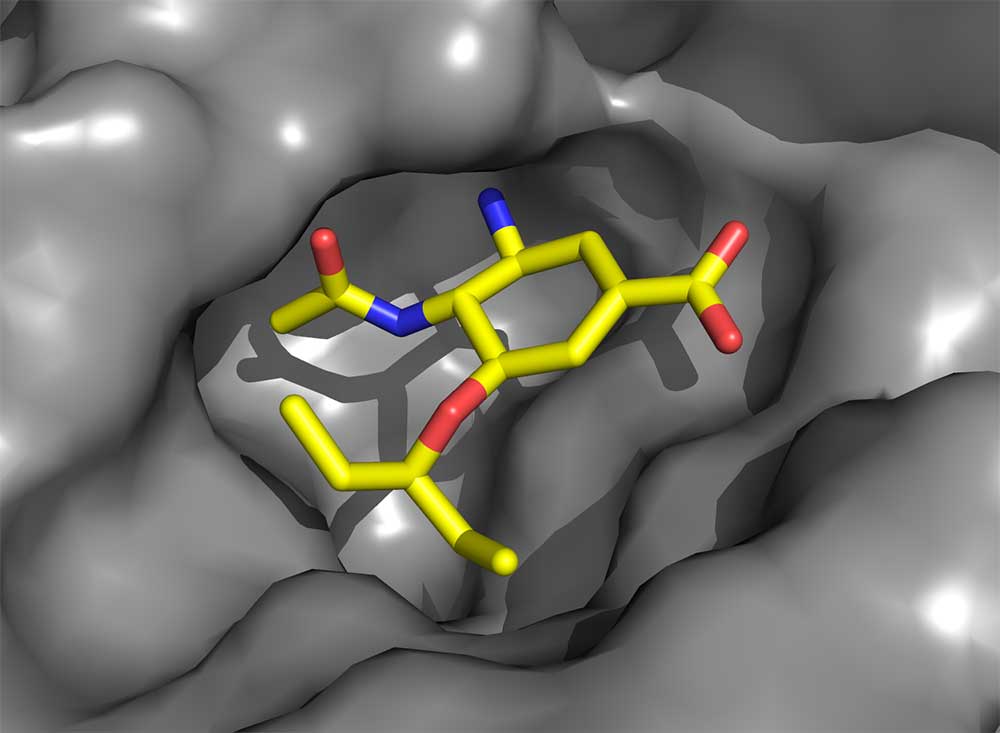
Oseltamivir carboxylate has many of the features of zanamivir, but critically important changes were made, not to enhance its affinity but to improve its pharmacological properties to allow oral administration. These changes come with some cost because now the neuraminidase structure needs to change very slightly (of the order of an atomic diameter or so) in order to bind the drug. In 2000, we observed that this structural change introduced a ‘resistance window’ that could compromise Tamiflu’s capacity to work against all virus strains. Resistant viruses are readily selected in the laboratory simply by culturing the virus in the presence of the drugs. Later, a circulating influenza virus was discovered containing such a mutation (the substitution of amino acid histidine 275 with tyrosine, H275Y) that impeded the structural change required for oseltamivir carboxylate (but not zanamivir) to bind, and which was, as a result, resistant to Tamiflu.
Usually, influenza is an acute infection and drug treatment regimens are for only five days. Under these circumstances, the virus has little time to acquire resistance in a patient undergoing drug treatment. For Tamiflu, five years of monitoring such acquired resistance in patients infected with influenza A viruses resulted in a reported 5% incidence of resistance in viruses of the 2009 H1N1 pandemic strain (‘swine flu’) and a 2% incidence in H3N2 viruses. All of the N1 resistance was identified as H275Y, and all of the N2 resistance was R292K. For Relenza, which is far less-widely used, no comparable data is available.
Antiviral and antibacterial medicines all face limited utility depending on how quickly the virus or bacterium acquires resistance. One way to tip the odds in favour of the drug is to have it resemble the natural ligands of the target as closely as possible. Other ways include using cocktails of drugs that require multiple mutations before resistance emerges.
Other drug targets in the influenza virus are being actively pursued, including the proteins that copy the viral genes. Typically, these research endeavours are headlined with the promise of drugs that will be effective against all influenza strains. Lessons learned from the neuraminidase inhibitors should inform considerations about the likely emergence of drug-resistant viruses and aid clinical trial design to improve the likelihood of establishing bone fide clinical efficacy.