On the night of 10 December 2018 in Stockholm Sweden, half of the Nobel Prize in Chemistry was awarded to Frances H. Arnold (Caltech, USA) ‘for the directed evolution of enzymes’ and the other half was shared between George P. Smith (University of Missouri, USA) and Sir Gregory P. Winter (Cambridge, UK) ‘for the phage display of peptides and antibodies’. Arnold was the first to artificially direct the evolution of enzymes and Smith and Winter developed ‘phage display’ for the directed evolution of antibodies, both harnessing the power of evolution for the benefit of chemistry.
Directed evolution of catalysis and affinity
In biology, most proteins perform either a catalytic (e.g. enzymes) or an affinity (e.g. antibodies, receptors, carrier proteins) role. The proteins bind to either a ground state (affinity) or a transition state (enzyme) of another molecule, but it is essentially a recognition process. All these proteins have been shaped by evolution over the past 3.7 billion years (or so). In fact, there is no better place to see evolution at work than in the protein sequences of ubiquitous proteins such as the FK506 binding proteins (BMC Dev. Biol. 2018, vol. 18, p. 7). Gradual changes over geological timescales have led to all the species we see on Earth today. But, clearly, this process is too slow to produce catalysts for new reactions at the rates we need to find them to stoke the fires of industry or to find that magic cure for a sick relative.
The idea of ‘directed evolution’ of proteins was first proposed by Manfred Eigen as a theoretical idea. From 1953, he worked at the Max Planck Institute and became its Director in 1964. He received a Nobel Prize in Chemistry with Ronald Norrish and George Porter for the study of ultrafast reactions in 1967. The following year he turned his attention to the problem of self-organisation of matter and the evolution of biological macromolecules, and in 1984 proposed a method for the direct evolution of enzymes (Pure Appl. Chem. 1984, vol. 56, p. 967). He reasoned that it would be impossible to find improved variants of an enzyme in random libraries. The average protein has 250 amino acids, and if only 100 are ‘mutated’ that would be a library of 20100 members. So many, in fact, that there is not enough matter in the universe to make even one molecule of each. Instead, Eigen proposed several generations of mutagenesis and screening and predicted that it would be possible to evolve molecules to produce optimised enzymes.
An alternative, of course, is not to make the library but to model them in silico, as it were. This was proposed by a scientist at Genex Corp also in 1984 (World Biotech. Rep. 1984, vol. 2, p. 233) – Robert C. Ladner. Ladner went on to found Protein Engineering Corp. where he patented a way to facilitate the ‘directed evolution of novel binding proteins’ (US6979538B2) by encoding the library in bacteriophage and expressing these on their surface and then separating the binding phage and introducing errors in the DNA to evolve the best binding variants.
Frances H. Arnold

Niklas Elmehed. © Nobel Media
Frances Hamilton Arnold was born on 25 July 1956 in Pittsburgh, daughter of the famous nuclear physicist William Howard Arnold. She attended the city’s Taylor Allderdice High School and moved out of home by age 17, hitchhiked to Washington DC to protest the Vietnam War and skipped school to work as a taxi driver and as a cocktail waitress by night. She got into Princeton, not on her grades (which were average), but on near-perfect standardised test scores and a compelling essay explaining her lack of interest in the traditional high school curriculum. While at Princeton, she continued to drive cabs and made several solo trips to South America and Europe on the proceeds. She graduated in mechanical engineering (1979) as one of fewer than five women and then went to work for the Solar Energy Research Institute (SERI) in Colorado, envisaging a future free of fossil fuels.
However, the oil crisis finished, Americans drove big cars again and Frances’ brother (Eddy Arnold, professor of chemistry at Rutgers) encouraged her to ‘try chemistry’ instead of solar research. So she left SERI in 1980 destined for a PhD in chemical engineering at Berkeley. Just in time, as it turned out. A year later Ronald Reagan cut SERI’s budget by 90% and almost everyone there lost their job.
At Berkeley, she realised that she didn’t know any chemistry and had to take the entire undergraduate curriculum, which she completed in 12 months, and then began working on biofuels with Harvey Blanch. Reagan’s cuts also saw Blanch’s funding for biofuels cut to zero and so he moved into biotechnology.
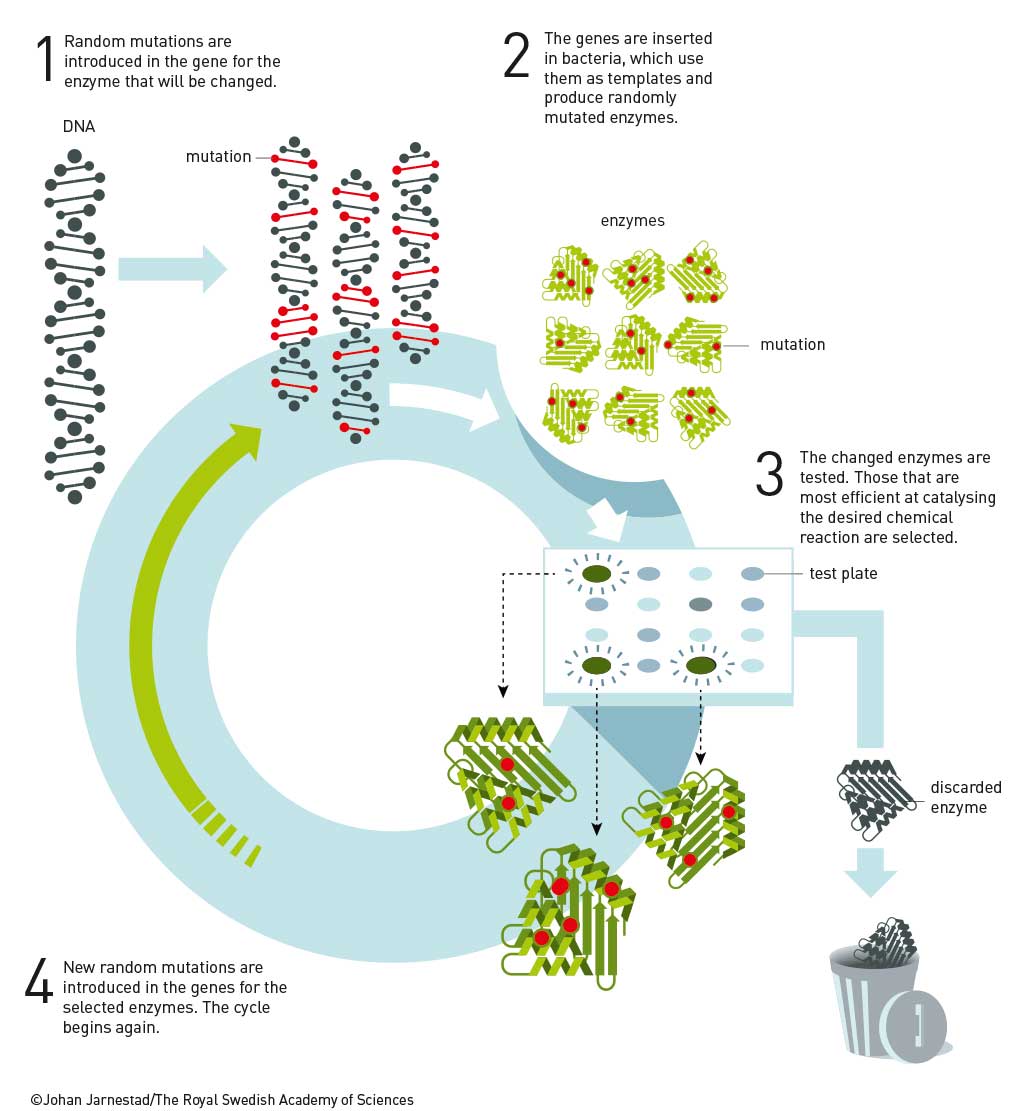
Arnold finally obtained her PhD in 1985 on affinity chromatography. She continued to work for 18 months at Berkeley for Ignacio Tinoco until obtaining a position at CalTech in late 1986. While at Caltech she brought Manfred Eigen’s vision to a technically elegant reality. Arnold developed rapid iterative methods that mimicked the slow random mutagenesis and selection found in nature. Coupled with innovative screening techniques to select mutations that addressed targeted properties, this resulted in the discovery of enzymes that worked in organic solvents (Proc. Nat. Acad. Sci. USA 1993, vol. 90, p. 5618), enzymes that catalyse reactions invented by chemists and not known in nature, such as olefin cyclopropanation (Science 2013, vol. 339, p. 307), and enzymes that catalyse the formation of bonds not found in nature, such as C−Si bonds, through carbene insertion into Si–H bonds (Science 2016, vol. 354, p. 1048). C−Si bonds are useful in medicinal chemistry and a wide variety of consumer products, but are unknown in biological systems.
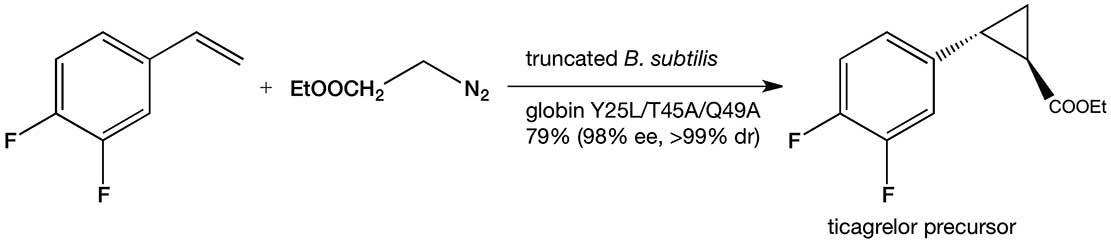
The discovery of enzymatic olefin cyclopropanation started with a literature report of haem mimics performing cyclopropanation in organic solvents. Screening of haem proteins for this activity led to the discovery of a bacterial cytochrome P450 that displayed a low level of promiscuous (cyclopropanation) activity when fed diazocarbenes and a suitable olefin. Directed evolution involved DNA sequence library construction, mutating residues in or near the active site. This step is critical and relies on a detailed knowledge of the enzyme’s structure. Not unexpectedly, the full sequence space for any enzyme is very sparsely populated in activity, which tends to be clustered in a few islands. The trick is to stay close to land. In the next step, screening of the library for the reaction you want allows you to narrow the field and re-mutate the most active variants from the first screen and then repeat the process. In just a few generations of directed evolution, the Arnold group was able to improve the activity and selectivity of the enzyme so that it could produce, for example, a precursor for the heart drug ticagrelor with very high diastereo-and enantio-selectivity (see diagram below).
Another aspect of this work is to replace toxic heavy metals with green enzyme catalysis. For example, the industrial synthesis of sitagliptin, a selective DPP-4 inhibitor for the treatment of type 2 diabetes, is made via a key step that requires RhI/t-Bu JOSIPHOS, an expensive heavy-metal-containing catalyst. The scientists at Merck were able to use 11 rounds of enzyme evolution on an artificial (R)-selective transaminase to produce an enzyme that did the same reaction in much higher enantioselectivity and without the use of toxic heavy metals.
Another way to navigate the sparse activity landscape of enzymes, and stay close to land, is to use related enzymes to guide mutations. This is called ‘DNA shuffling’ and was introduced by Pim Stemmer in 1994 (Nature 1994, vol. 370, p. 389). In this process, pieces of DNA for the same enzyme in several organisms are shuffled like a deck of cards. In this way, mutations are directed toward areas already known to produce active enzymes. The same process has been used for antibodies (called affinity maturation), and is much more efficient than random mutations at accessing wider chemical space. These methods have been facilitated by the advent of cheap DNA synthesis and sequencing and, it could be argued, would not have been possible without this (Gilbert and Sanger, Nobel Prize in Chemistry 1980; Ochoa and Kornberg, Nobel Prize in Physiology or Medicine 1959).
The other half of the Nobel Prize went to the directed evolution of ‘affinity’ (as opposed to ‘activity’), which is facilitated by phage display, a method of physically coupling a phenotype (high-affinity binding protein) with a genotype (the DNA for that protein). Phage, or more properly ‘bacteriophage’ are the champions of the underdog. In the wild, when any species of bacteria threatens to take over a local environment, their viruses (phages) come in to decimate the Johnny-come-lately. With an estimated global population of 1031 and launching 1024 successful infections per second, they have major effects on the global energy and nutrient cycles – much of biology is about feeding the phages. They were also crucial to identifying DNA as the genetic material in 1952 (Alfred Hershey, Nobel Prize 1969), that DNA had a triplet code in 1961 (Francis Crick (Nobel Prize 1962), Leslie Barnett, Sydney Brenner (Nobel Prize 2002), and Richard Watts-Tobin) and later in uncovering the mechanism of gene regulation, protein folding, DNA binding and genetic recombination. Look anywhere in molecular biology and you will find indispensable phage enzymes at work.
George P. Smith

Niklas Elmehed. © Nobel Media
George Smith was born 10 March 1941 in Norwalk, Connecticut (USA), and obtained a BA in biology from Haverford College in 1961. He was fascinated by reptiles and hoped to become a herpetologist, but his mathematical way of thinking made him much more suited to molecular biology. His senior thesis project tried to answer the question of how the immune system can recognise anything foreign. His experiments were naive and unsuccessful and actually solved the next year by Edgar Haber (Harvard). After a year as a high school teacher and lab technician, Smith was accepted into a PhD program at Harvard with none other than Edgar Haber, who was also hosting another (future) Nobel laureate at about the same time (Bob Lefkowitz). After a postdoc at Wisconsin with another future Nobel laureate (Oliver Smithies), he joined the faculty at the University of Missouri in 1975 and has remained there till this day.
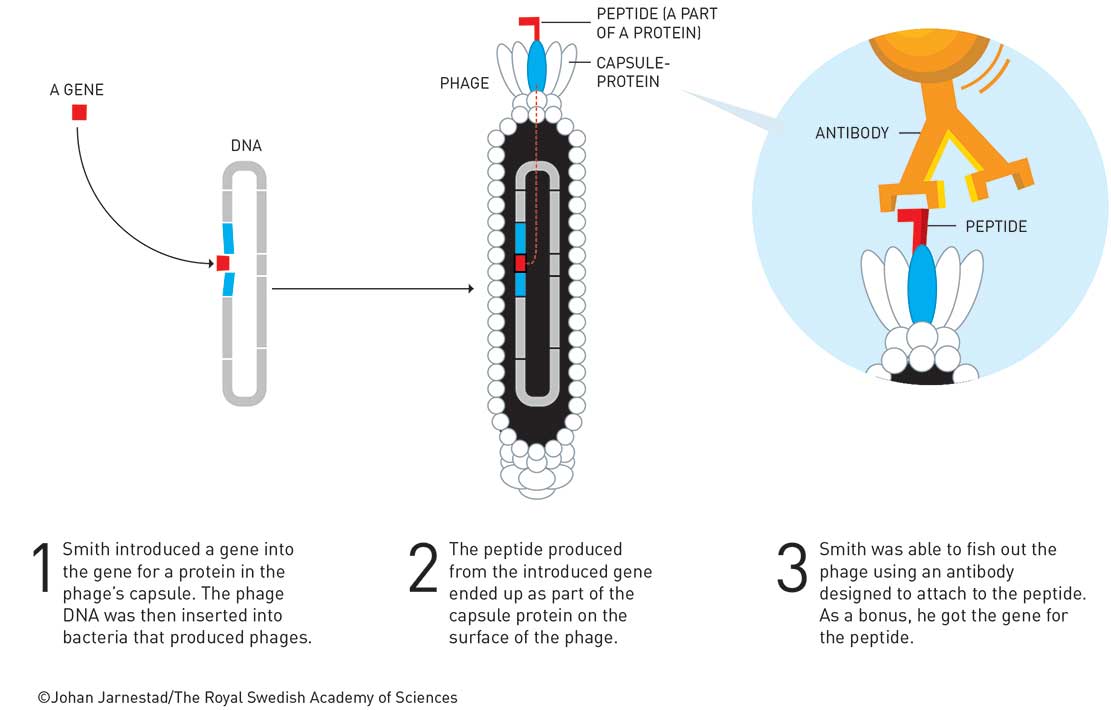
Smith has a career total of about 50 papers. He got none from his PhD but several as a postdoc at Wisconsin, including in Science and Cell. Notably, his first paper as an assistant professor at the University of Missouri was also in Science – a sole author paper about the evolution of repeated DNA sequences. He became interested in phage and using these as cloning vectors. For example, he discovered a new filamentous phage cloning vector (Gene 1980, vol. 9, p. 127) in 1980. Here he was able to clone the tetracycline resistance gene into the fd phage and show that infected E. coli became tetracycline resistant. However, it was a two-year sabbatical (1983–4) at Duke University with Robert Webster that led to the Nobel-winning breakthrough. Webster recalls that Smith was able to demonstrate that it was possible to fuse a foreign protein to the pIII coat protein of phage fd (Science 1985, vol. 228, p. 1315). The chimeric protein pIII-EcoR1 was able to be packaged into the phage and was correctly displayed on the surface so that an EcoR1 antibody could be used to purify the phages displaying the enzyme by 108-fold in three rounds of ‘biopanning’. Smith coined the term for affinity purification of phages displaying EcoR1 from a background wild-type phage, using a streptavidin-coated Petri dish to which biotinylated EcoR1 antibody was adsorbed (Gene, 1988, vol. 73, p. 305). Coincidentally, the restriction enzyme EcoR1 and the antibody used by Smith to confirm the enzyme was functionally expressed on the surface of the phage particle were supplied by Paul Modrich (Nobel Prize in Chemistry 2015, for DNA repair) also at Duke. Smith immediately recognised the power of this and advanced the technique over the years. By 1990, he had published the first ‘phage display’ library. This library was composed of peptides, ‘displayed’ on the surface of filamentous phage particle; a different peptide for every phage particle as encoded by the DNA inside (40 million hexamers in this case). He was able to purify specific phages that bound to an immobilised antibody (Science 1990, vol. 249, p. 386).
Of course, there were many scientists working on phage at this time and in many ways Smith just happened to be in the right place at the right time. On the other side of the Atlantic, Sir Gregory P. Winter reported the display of a folded and fully functional antibody fragment on filamentous phage also in 1990. This was something new and exciting because, for the first time, it decoupled an antibody from the animal that produced it.
Gregory P. Winter

Niklas Elmehed. © Nobel Media
Greg Winter was born six weeks prematurely in Leicester, UK, in 1951, and was a sickly child. His parents moved to West Africa (Legon in Ghana), where Winter grew up, in part, to keep young Gregory warm, which was near impossible in post-war London. He thrived in Africa (despite contracting malaria and some other tropical diseases) and recalls that what launched him into science was the sight of a huge sea turtle brought to school by the first ‘scientist’ he had ever met. Gradually he discovered that the unknown frontiers he craved were not so much of the David Attenborough sort but those associated with the unseeable molecular world. Winter returned to the UK for high school at the Royal Grammar School, Newcastle upon Tyne. He went on to read natural sciences at the University of Cambridge, graduating from Trinity College, in 1973 (of which he became Master in 2012). Staying on at Cambridge, he was awarded a PhD in 1977 from the MRC, for studying tryptophanyl-tRNA synthetase from Bacillus stearothermophilus. He was supervised by Brian S. Hartley and George Brownlee and, unlike Smith, wrote up five papers from his PhD. He left Cambridge (briefly) for a postdoc at Imperial College and then another at the Institute of Genetics at Cambridge before becoming a group leader at the MRC in 1981.
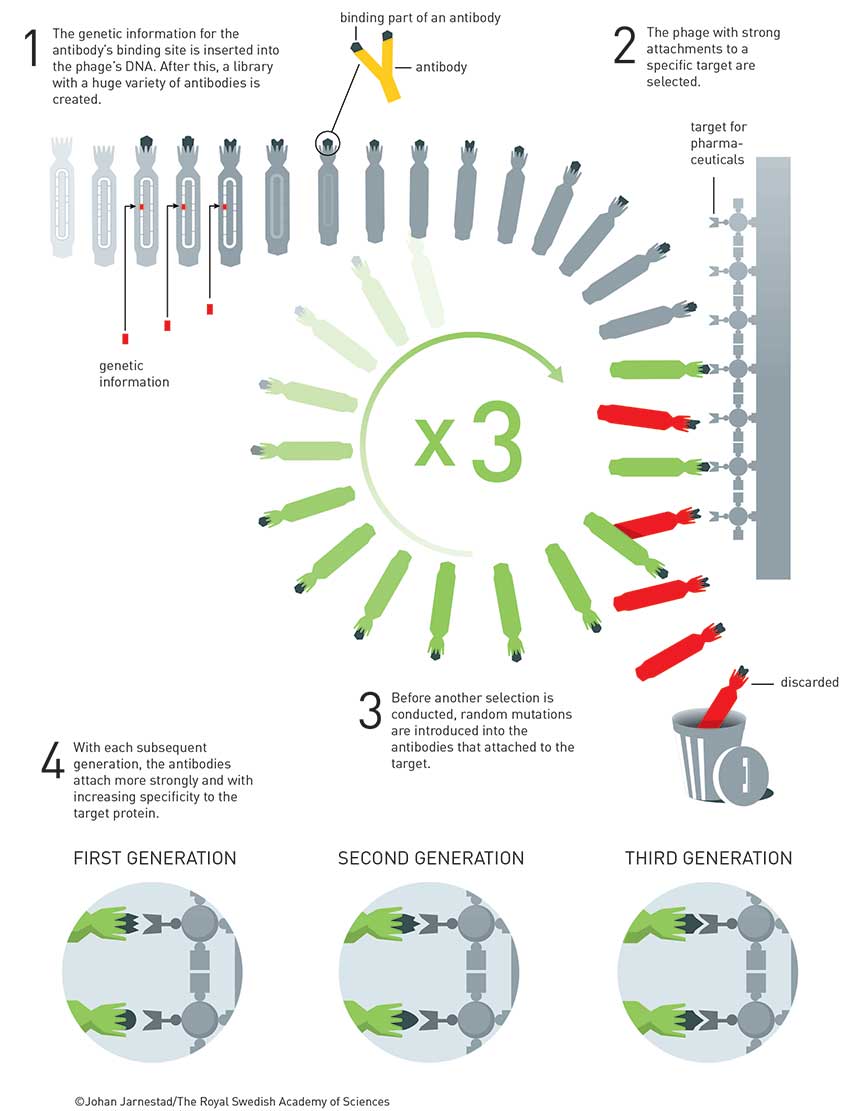
His first encounter with phage was in 1980 (Nucl. Acids Res. 1980, vol. 8, p. 1965), when he cloned influenza cDNA into bacteriophage M13. In 1982, he won a fellowship and travelled to Canada to pursue work on how mutations affected enzyme function. On his return to the UK, César Milstein (Nobel Prize 1984) suggested that Winter should work on antibodies, the development of which for human use was hampered because they were raised in animals, typically mice, so contained too much ‘mouse’. What was needed to develop antibody drugs was a way to ‘humanise’ them so they would not produce an immune response. Winter responded with intense excitement about the possibilities of using site-directed mutagenesis to alter antibody structures. He postulated that antibodies were just scaffolds with only small sections required for antigen recognition.
He initially worked with Herman Waldmann, who was using antibodies raised in rats to treat leukaemia patients, and Sir Alan Fersht, who was a protein engineer. Together, they made a major leap forward by replacing large sections of rat antibody with the human equivalent and found the antibodies still worked as before. At about the same time, Winter was also using M13 phage display to dissect the tyrosyl-tRNA synthetase enzyme to see what sections were required and which were not. Applying this technique to humanising antibodies was a logical step forward but one Winter was also quick to realise had huge commercial potential.
In 1989, he set up Cambridge Antibody Technology (CAT) to commercialise this work. In the first paper, Winter took an scFv (single-chain variable fragment of an antibody) derived from mice immunised against chicken egg-white lysozyme and fused it to protein pIII of fd phage, taking inspiration from Smith’s 1985 paper. They showed that the scFv-phages had high specificity and could be amplified 106 times over other phages in two rounds of affinity purification (Nature 1990, vol. 348, p. 552).
In collaboration with BASF, CAT developed adalimumab, which was bought by AbbVie and marketed as Humira. It was the first fully human antibody (against TNFα) drug ever developed by using phage display and the world’s largest selling drug in 2017 (US$18 billion). It is used to treat inflammatory diseases such as rheumatoid arthritis and Crohn’s disease and costs about US$52 000 per year in the USA. It came off patent in 2018 but biosimilars appeared in India in 2014 and are now globally available at US$2400 per year.
The first biologic drug (chemically humanised insulin) became available in 1982, and by 2016, there were 64 fully human antibody drugs that had been developed by phage display in the clinic or in clinical trials. This may well only be the vanguard and the ensuing decades may see small molecules largely replaced with biologics, or the two drug discovery paradigms may merge as is being witnessed by the emergence of antibody–drug conjugates. In late 2018, there were four conjugates approved – brentuximab vedotin (Seattle Genetics), ado-trastuzumab entansine (Genetech/Roche) inotuzumab ozogamicin (Wyeth/Pfizer) and gemtuzumab ozogamicin (Wyeth/Pfizer).
Winter went on to found his second (Domantis in 2000) and third company (Bicycle Therapeutics in 2009) but still remained first and foremost an academic who thinks about practical problems. His thesis advisor once told him ‘Bugger interesting, is it an important question?’ Virtually the same advice was given to Fraser Stoddard by his PhD advisor at their second meeting (at his thesis defence).
The 2018 Nobel Prize in Chemistry recognises Arnold, Smith and Winter, all with very different trajectories, for ‘hacking’ evolution to generate better drug treatments faster, greener and more efficient chemical manufacturing processes and more economical biofuels. Thanks to these innovations, what nature takes millennia to do, chemists can achieve in weeks or months. I think, if nothing else, this year’s Nobel Prize provides final proof for the molecular basis for Darwin’s theory of evolution.